




{kind=link}
This is a part two of a series of blog posts about using Dataiku in biotech (you can check out the first post in the series about parsing Variant Call Format files here). In this article, we'll show you how to sequence DNA with RNA-Seq. * Note: This blog post contains screen shots from an older version of Dataiku, though all of the functionality described still exists in the latest version. Watch the on-demand demo to take a look at the latest release of Dataiku in action.
For the past decade, the price of sequencing a human genome has declined exponentially, and it's already affecting the way doctors treat their patients. Sequenome's MaterniT21 test is rapidly replacing aminocentesis as the safest way to diagnosis Down's syndrome, and Oncotype DX gives personalized cancer prognoses based on genomic data. Plus, direct-to-consumer companies, like 23andMe and Ancestry.com, could further alter the doctor-patient relationship (if they can solve their FDA troubles).
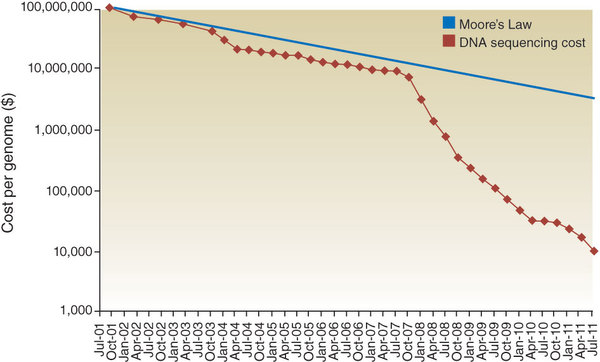
In short, the future of many biotech companies is going to depend on leveraging genomic data. We're in the era of precision medicine. It's now possible to optimize clinical trials with exactly the right patient population, rapidly discover new biomarkers and give accurate prognoses to patients. But before biotech and pharmaceutical companies can start doing this, they need to be able to handle large volumes of sequencing data.
What's RNA-seq?
One of the most popular sequencing technologies is RNA-seq. It measures the expression - i.e., the amount of RNA that is created from DNA - of each gene in the genome. RNA-seq was invented less than five years ago, but it has already been used in thousands of biomedical studies. Researchers know that correctly interpreting this data is key to optimizing clinical trials and discovering novel biomarkers.
However, RNA-seq presents an enormous challenge for data processing. A sequencing machine produces terabytes of raw data known as reads - sequences of about 100 letters which correspond to a portion of the genome. In order to analyze this data, researchers have map those reads onto genes. In computer science, this is known as fuzzy matching, but biologists have to do this billions of times with terabytes of data.
I'm going to walk through this process in this blog post using Dataiku. Once we're done, we're going to have clean data ready for analysis and a data pipeline that can be reused on new data.
Getting Data into Dataiku
For an RNA-seq analysis, you need three input datasets:
- Your reads from a sequencing machine
- A reference genome
- The location of genes on the genome
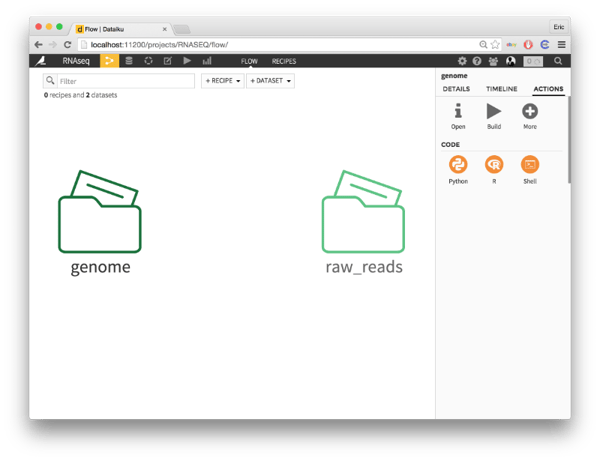
Building the Reference Genome
The first step is to take the mouse genome provided by UCSC and turn that into an index. This is a binary file, which you can use to do lightining-fast alignment using bowtie2.
In Dataiku, I clicked on my reference genome folder and then the shell widget on the right. This bring up an editor where I can compose a bash script. Importantly, if you click on the variables tab, you can see the environmental variables that Dataiku sets for the job. These are going to be key for the bash script.
Our bash script is only one line.
bowtie2-build $DKU_INPUT_0_FOLDER_PATH/refMrna.fa $DKU_OUTPUT_0_FOLDER_PATH/genome
That line is a bit confusing, especially for people who aren't used to bash, so let's go through it step-by-step. Dataiku creates several environmental variables that you can access from your bash script. $DKU_INPUT_0_FOLDER_PATH and $DKU_OUTPUT_0_FOLDER_PATH have the paths to the input and output folders of the script. So, we're passing bowtie-build the location of the input exome with $DKU_INPUT_0_FOLDER_PATH/genome.fa and telling it to put the compiled exome at $DKU_OUTPUT_0_FOLDER_PATH/genome.
We when return to the flow, we see our two original datasets and now a bash script creating the indexed reference exome:
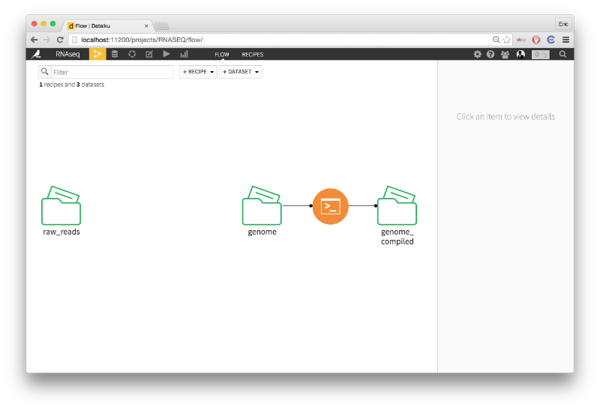
Aligning Reads
Next up, we have to take the raw reads from the sequencing machine and align them against the compiled genome. I'm using tophat2 to do this. In Dataiku, I created a new bash recipe. I specify two inputs from the recipe - the indexed reference genome and the raw reads - and one output folder, which I'll call mapped reads. The resulting script is itself just one line.
tophat2 -o $DKU_OUTPUT_0_FOLDER_PATH \
-G /path/to/known_genes.gtf \
$DKU_INPUT_0_FOLDER_PATH/genome \
$DKU_INPUT_1_FOLDER_PATH/raw_reads.fastq
Going step-by-step, we first specify the output directory for the mapped reads with $DKU_OUTPUT_0_FOLDER_PATH. Then, we specify the location of the file with the location of known genes. Then, we specify the compiled exome with $DKU_INPUT_0_FOLDER_PATH/genome and the mapped reads with $DKU_INPUT_1_FOLDER_PATH/raw_reads.fastq.
Returning to the flow, we see a new folder appear:
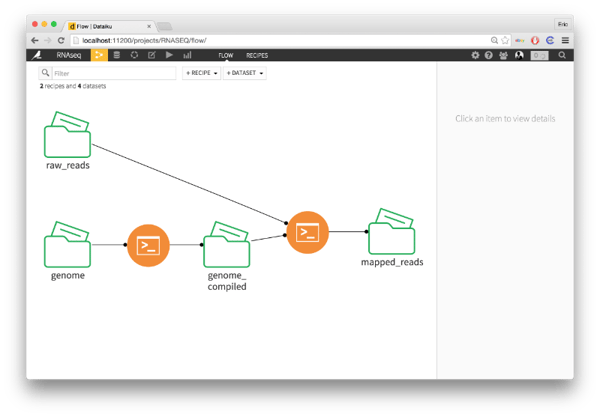
Counting Reads per Gene
The last step is to take the aligned reads and count up the number of reads for each gene. Here, I'm using cufflinks. This time, I'm going to specify the output as a dataset rather than a folder, because the output of cufflinks is a nice tabular dataset. Our bash script is again one easy line:
cufflinks $DKU_INPUT_0_FOLDER_PATH/accepted_hits.bam
And our flow gets one more dataset.
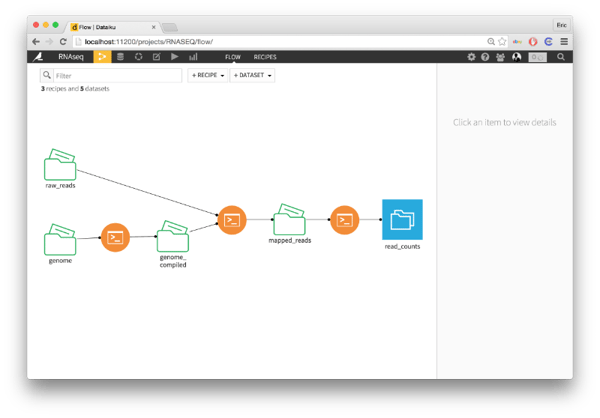
Great! Now when have our entire flow. If we click on the final folder and click build Dataiku will execute all the steps to perform the read alignment. Of course, there are tons of parameters that you could change for each of these steps. You could, for instance, use multiple cores for each processing step with the -p option, or maybe you could distribute the computation across a grid engine for even faster computation. But I just kept this tutorial simple to give you the basics
Conclusion
In a few minutes, we created a simple RNA-seq processing pipeline in Dataiku. The final dataset can then be analyzed in Dataiku using R, Python, SQL or our visual analysis tools. You could even use Dataiku to create a web-based visualization of the results.