




{kind=link}
The past decade has seen a dramatic fall in the price of a human genome, and there are amazing open-source databases filled with genomic information, so anyone can access terabytes of genomic data. We'll use this to build a predictive model of breast cancer metastasis with Dataiku DSS.
I'm going to look at one of these databases: The Cancer Genome Atlas. Specifically, I'm going to look at the mutations that happen in breast cancer. I'm going to do two specific things with this data. First, I'm going to run a clustering algorithm on the cancers to see if there are different types of breast cancer. Then, I'm going to build a predictive model for cancer recurrence, otherwise known as metastasis.
*Note: This blog post contains screenshots from an older version of Dataiku, though all of the functionality described still exists. Watch the on-demand demo to take a look at the latest release of Dataiku in action.
Data Preparation
The data that I'm working comes in two files. I have a .csv file with the 200 most common mutations that occur in breast cancer. Patients are represented by rows in the dataset, while the columns represent mutations with either "true" or "false" if the mutation is present. I also have clinical information on each of the patients: their treatments, when they were diagnosed, their outcomes, etc.
Clustering Cancers Based on Genetics
First, I wanted to cluster the cancers based on their mutations to see if there are genetically distinct types of cancers. Doing this is super easy with Dataiku. I clicked on the common_mutations dataset, which brings up the data explorer. Then, I clicked on Models.
Right now, I just want to explore the dataset with clustering, so I clicked on clustering to do so.
Dataiku automatically chooses an appropriate clustering algorithm. For this dataset, it went with k-means clustering, which is a super commonly used method. You can go into settings to choose from several different algorithms.
Dataiku also produces a summary of each of the clusters, which is really interesting for this particular data set. We see three large clusters: cluster0, cluster2 and cluster4. Clicking on cluster0 brings up a summary.
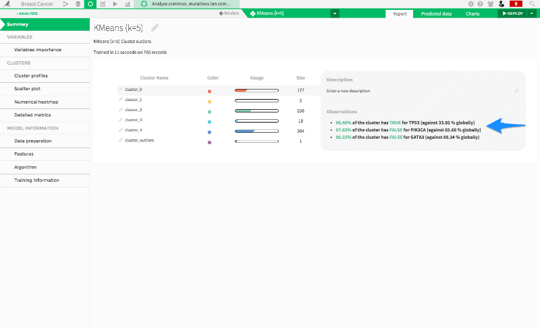
We see that cancers in this cluster at almost all positive for TP53 mutations, while almost all of them are negative for PIK3CA mutations. These are two genes that are known to cause cancer, also known as oncogenes.
In contrast, we can look at cluster_2:
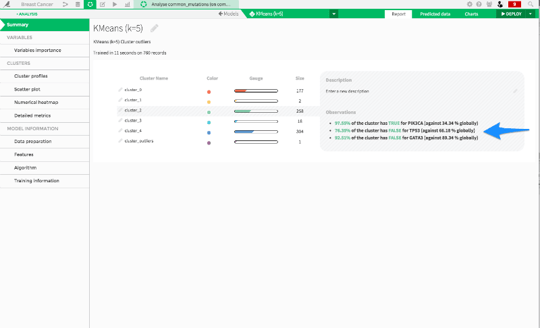
These cancers are almost all negative for TP53 mutations, while they are almost all positive for PIK3CA mutations. This is the exact opposite of cluster_0!
Finally, we can look at cluster_4:
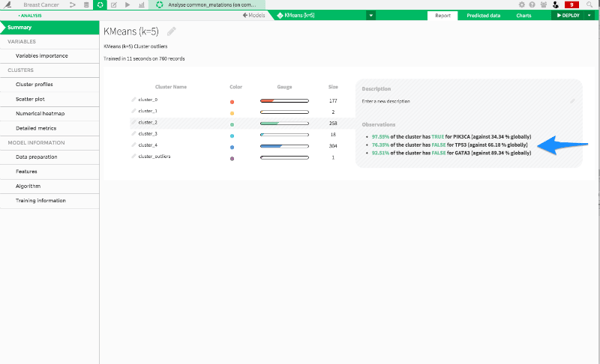
This cluster is negative for both TP53 and PIK3CA mutations. These are the remaining cancers that don't fit into the two other categories. So, with just a few clicks, we were able run a clustering analysis on hundreds of cancers. We found three main types of cancer: those caused by TP53 mutations, those caused by PIK3CA mutations, and then a group of remaining cancers. Now, let's do some predictive modeling!
Predictive Modeling of Metastasis
I'm most interested in building a predictive model of cancer recurrence. In general, having a recurrence is really bad for a cancer patient, so clinicians are often most interested in predicting this variable.
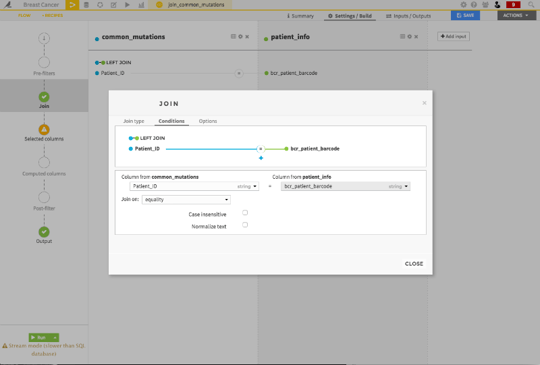
In order to build this model, I need to join the table containing the mutations with the table containing the patients' clinical information. Fortunately, joins are really easy with Dataiku. To do a join, I click on the mutation dataset and then the join widget on the right of the screen. This will up a dialog box where I can choose the second dataset.
Then, Dataiku wants you to specify the column which pairs rows in one dataset to rows in the other dataset. In this case, it was the identifier for each patient.
I can then select the columns from the two datasets that I want to include in my two new datasets.
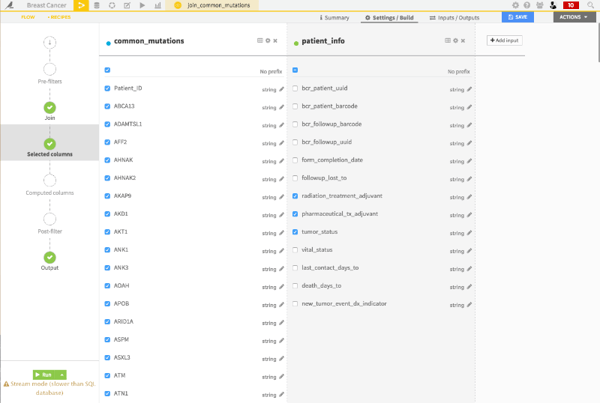
Awesome. Now, I have a third joined dataset, which combines two initial datasets.
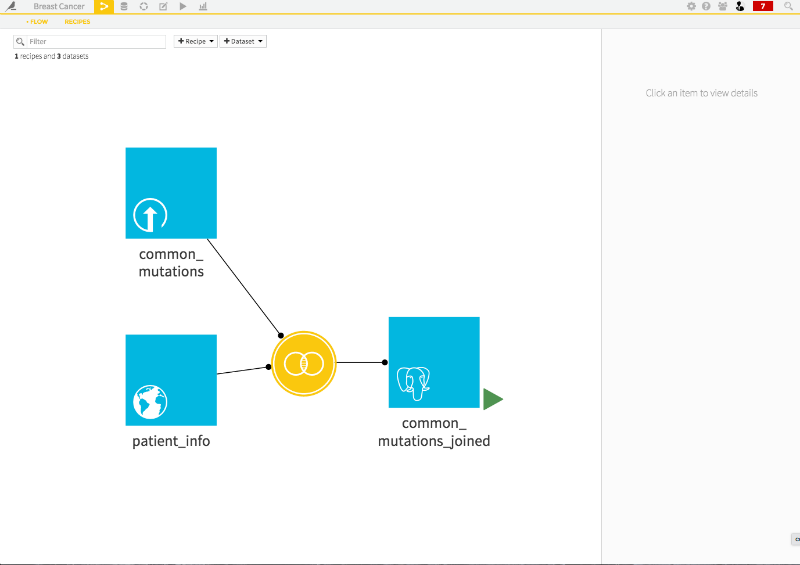
Clicking on this new dataset and then the analyze widget, brings up the explorer. I'm interested in predicting the patients' tumor status, so I click on the tumor_status column and then create prediction model.
Dataiku automatically trains three separate models: a random forest, a support vector machine (SVM), and a logistic regression model.
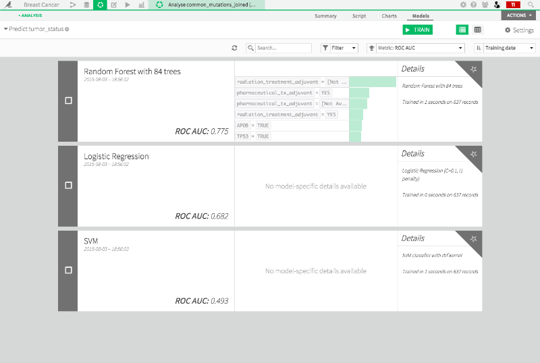
Dataiku evaluates models according to AUC. All you really need to know is that higher AUCs indicate better performance, and an AUC of 0.80 is generally what's considered necessary for a model to be clinically useful. We see that the random forest performed the best and is at 0.775, pretty close to the 0.80 cutoff for a model to be clinically useful. Let's click on that model to explore it a bit.
Dataiku automatically calculates the importance of the variables used in the predictive model.
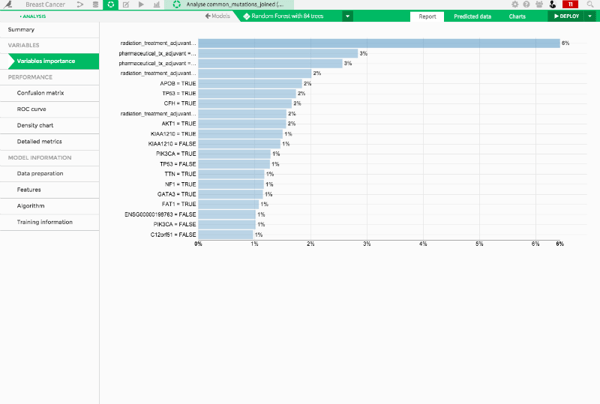
According to variable importance, we see that radiation treatment and pharmaceutical treatment are the two most important variables for predicting whether a patient has a metastasis. After those clinical variables, we have APOB and TP53, which are both genes known to be related to breast cancer.
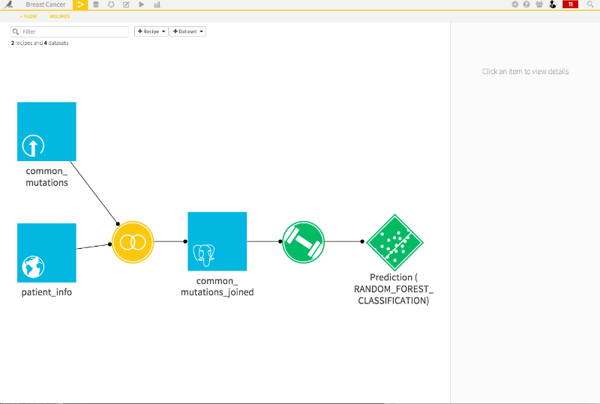
Let's go ahead and deploy this model to our flow. Once the model is deployed, we can apply the model to new datasets and automatically retrain the model if input datasets change. After clicking on Deploy in the upper right screen, I can go back to the flow and see the deployed model.
Recap
That's it! In just a few minutes, we've clustered our samples to find three major genetically distinct types of breast cancer. Then, we joined the mutation data with information about the patients and built a random forest to predict metastasis. If you thought this was interesting, you can check out more of our blog posts about creating web apps (like this one predicting the crime rates in London) or try out Dataiku on one of your own projects!